ICME 2015 HW1
Contents |
Overview
This homework takes place at the electronic scale and is separated into two parts:
- Density Functional Theory (DFT)
- Interatomic Modified Embedded Atom Method (MEAM) potential development
All necessary input files and scripts are provided in the /cavs/general/Projects/ICME_2015/HW1/ directory. Move these files to your own directory (and make a backup copy) before trying to perform any simulations.
Use /scratch/"Your Directory" for best results.
Write a full report that follows a journal article manuscript format (include figures and tables in the text).
Upon completion, upload a .pdf and .doc(x) file to your group folder in the ../ICME_2015/HW1/ directory. Be sure to also upload the requested files and plots from each section of the homework.
Part 1 - DFT Calculations
For this course, use Vienna Ab initio Software Package (VASP) to run DFT calculations.
Assignment Sections
Energy-Volume (EV) curves (upscaling for MEAM calibration)
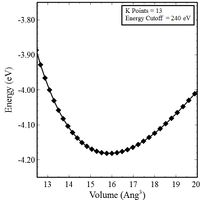
1. Plot energy (E) vs. volume (V) and energy (E) vs. atomic separation (A) curves for nickel in a FCC crystal structure (set PREC = Normal).
2. For the FCC crystal structure, perform a KPOINT convergence study (set PREC = Normal). Choose the KPOINT grid that reaches a converged solution (think of this as a mesh refinement).
- a. Plot lattice parameter vs. KPOINT grid
- b. Plot bulk modulus vs. KPOINT grid
- c. Plot cpu time vs. KPOINT grid
3. Provide a thorough analysis of the four (4) equations of state (EOS) used for deriving the equilibrium lattice constant, bulk modulus, and cohesive energy e.g. How does changing the range of the simulation change the derived parameters?
4. Come up with final converged KPOINT grid and plot final E-V and E-A curves (set PREC = High).
5. Provide a final set of parameters using the converged KPOINT and EOS (of your choice). This data will be use for calibrating the MEAM potential.
6. Report on your results
- a. Copy all summary files to your ICME group folder
- b. Copy final E-A curve to the ICME group folder
Properties of Nickel (upscaling for MEAM calibration)
Refer to guidelines http://cms.mpi.univie.ac.at/vasp/vasp/Surface_calculations.html .
Use the input files provided on the ICME website to calculate:
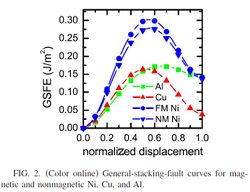
1. Using the input files provided on the ICME website produce a GSFE curve for nickel.
- a. Use the converged KPOINT value obtained from the previous section
2. Report your results
- a. Plot the change in energy vs. upper atom displacement
- b. Copy final GSFE curve to your group folder
Examples for performing the necessary simulations are found by clicking the on the respective image below:
 |
 |
|---|
Part 2 - MEAM Potential Development
For this portion of the assignment, use the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) package.
A comprehensive user manual can be found here.
Assignment Sections
MEAM Potential Calibration/Validation
Fit the modified embedded atom method (MEAM) model and generate a new MEAM potential for nickel by using the property values calculated above from the DFT results.
The calibration will include values calculated by DFT: Equilibrium nearest neighbor distance/lattice parameter, cohesive energy, and the elastic moduli (sometimes the surface formation energy for (111) surface is used but not in this homework). Elastic constants can be checked with literature results (experimental values in Table III page 15 of Mehl et al.).
For validation we will only examine the stacking fault energy although many other calculations are usually performed such as: vacancy formation energy, self-interstitial energy (octahedral and tetrahedral), etc. (see Jelinek et al., 2012).
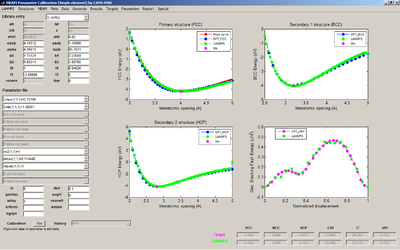
1. Using the MEAM Potential Calibration (MPC) routine determine the MEAM constants by comparing the energy versus atomic separation (E-A) plots from DFT and the Rose Equation:
- a. Using DFT results set alat, esub, and bulk
- b. If the Rose Equation results for alat, esub, and bulk do not fit well, modify the values to get a better fit
- c. Plot a comparison of the E-A curves for the initial alat, esub, and bulk and the optimized alat, esub, and bulk cases on one plot
- d. Compare the DFT values and Rose Equation optimized values
2. Using the MEAM Potential Calibration (MPC) routine determine the MEAM constants by comparing the energy versus atomic separation (E-A) plots from DFT and LAMMPS:
- a. Using DFT results set alat, esub, and bulk
- b. Calibrate the MEAM constants to the DFT results
- c. Plot a comparison of the E-A for the different cases on one plot
3. Conduct a validation check by comparing the MEAM stacking fault to the DFT result.
- a. Using DFT results for the GSFE calibrate the MEAM constants using Cmin, asub, b1 and b3
- b. Plot a comparison of the GSFE from DFT and GSFE from MPC
4. Sensitivity analysis
- a. Vary several of the parameters (one at a time) up to 50% to show the sensitivity of the parameters on the E-A curve and GSFE curve
- b. Plot the comparisons for E-A and GSFE curves for the different cases on one plot each (one containing the E-A sensitivities and one containing the GSFE sensitivities)
5. Report your results
- a. Place a screenshot of the final fitting routine and parameters in your group folder
6. Add a section or sections on improving the tutorial by adding/modifying the ICME webpage used for:
- a. VASP
- b. LAMMPS
 |
|---|
License
By using the codes provided here you accept the the Mississippi State University's license agreement. Please read the agreement carefully before usage.
Additional Guides
References
- ↑ C. Brandl, P. M. Derlet, and H. Van Swygenhoven, General-stacking-fault energies in highly strained metallic environments: Ab initio calculations, PHYSICAL REVIEW B 76, 054124 (2007)