Multiscale Modeling of Hydrogen Porosity Formation During Solidification of Al-H
Contents |
Abstract
This research proposal aims to model the formation and growth of pore in the liquid melt during the solidification of pure aluminum using a multiscale approach. The model will capture the effect of thermal gradient, cooling rate, melt temperature and initial hydrogen content on the growth kinetics and final porosity/pore size in solidified aluminum. To demonstrate the implementation of the proposed model, a case study on Al-H system is discussed. Application of the model to aluminum alloys should be straight forward.
Introduction

Aluminum casting offers economic advantages in reduced production steps and the ability to produce complex shapes since the metal is formed from the molten state. The specific weight of aluminum alloys makes them an attractive material in the automotive and aerospace industries where the need for fuel efficiency is ever present. However, the mechanical properties and performance of aluminum castings are strongly affected by structural defects, such as pores and entrained oxide films which degrade tensile strength, elongation, and fatigue life. Also, pores can lead to rejection of aluminum casting during final non-destructive inspection, such as X-ray, resulting in waste and an increase in production cost. Porosity resulting from metal casting can be divided into shrinkage pores and gas pores. Shrinkage porosity is due to the density difference between the liquid and the solid phases and occurs in sections of the casting that solidify later than the surrounding sections and do not have enough metal flow into the section for a complete fill. Shrinkage pores can be avoided by controlling the filling velocity. Gas porosity results from gas solubility difference between the liquid and the solid phase. Typically, the solubility of gas in higher in the liquid phase, during solidification, as the fraction of solid increases the excess gas in the liquid, supersaturates the melt, resulting in the formation of gas pores. In aluminum casting the main gas of interest is hydrogen. To quantify the final porosity that will result in a casted sample as a function of casting parameters such as thermal gradient, cooling rate, melt temperature, and initial hydrogen content, the nucleation, growth, and migration of pores must be currently modeled so that casting parameters can be adjusted accordingly to minimize porosity. This proposal will focus on understanding the mechanism of hydrogen porosity formation, works on shrinkage porosity will follow later. For the multiscale modeling Density functional theory (DFT) calculations will be performed for Al-H system to fit parameters in the modified embedded atom method MEAM potential. Molecular dynamics simulation using the MEAM potential will be done to obtain parameters needed for the phase field simulation of dendritic growth and pore nucleation and growth. The predicted porosity percent, average pore size, and pore morphology will be used in the Mississippi State University (MSU) Internal Stave Variable model to predict the mechanical properties (such as yield, ultimate, and fatigue strength) of the cast. Figure 1 illustrates this multiscale approach.
Methodology
Density functional theory (DFT)
The DFT calculates will provide the lattice parameters, the stable configuration, and other parameters to calibrate the MEAM potential for Al-H system. Care must be taken during the calibration stage to get the melting temperature, thermal expansion, hydrogen diffusivity and solubility in liquid and solid Al right.
Molecular dynamics simulation
Molecular dynamics simulations using the calibrated MEAM potential will be performed to calculate the kinetic coefficient of the solid phase, interfacial energy between liquid, solid, and gas phases, the energy of the system as a function of hydrogen content and temperature. These parameters will be upscaled into the phase field simulation to model the formation of hydrogen pore.
Phase field modeling
The system under consideration is a three-phase system ( ), represented by non-conserved phase variables
), represented by non-conserved phase variables  , and two components
, and two components 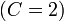 conserved concentration variables
conserved concentration variables  . The toal free energy,
. The toal free energy, ![F[J]](../images/math/6/1/d/61dbb08082cf1e0c5cf6b465458f62fe.png) of the system is composed of the chemical bulk energy
of the system is composed of the chemical bulk energy  and the interfacial energy
and the interfacial energy  terms:
terms:

The thin-interface approach for multi-component systems described in [1] is adopted in this work, where the interfaces are treated as a mixture of multiple phases, each with its own phase composition  and composition-dependent free energy function
and composition-dependent free energy function  . Therefore, the bulk free energy can be constructed using the interpolation function (
. Therefore, the bulk free energy can be constructed using the interpolation function (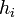 ) as,
) as,

where ![f_i[Jm^{-3}]](../images/math/4/f/7/4f7a3333067e85a8b3eac0961d1cc8dd.png) is the free energy density of phase
is the free energy density of phase  , calculated as the molar Gibbs free energy divided by the molar volume
, calculated as the molar Gibbs free energy divided by the molar volume  . The interpolation function introduced by [2] is used.
. The interpolation function introduced by [2] is used.

The multi-order parameter model presented by [3] is adopted and the interfacial term of the free energy is given by

Phase evolution equation
The evolution equation for each non-conserved phase field variable  is governed by the Allen-Cahn[4] equation:
is governed by the Allen-Cahn[4] equation:

where,

Another parameter needed from the molecular dynamics simulation is the interfacial energy of each phase which is related to  in the phase field (Eq.6) above. Also, the mobility of each phase,
in the phase field (Eq.6) above. Also, the mobility of each phase,  (Eq.5) of each phase can be calculated from the kinetic coefficient, which will be gotten from molecular dynamics simulation.
(Eq.5) of each phase can be calculated from the kinetic coefficient, which will be gotten from molecular dynamics simulation.
The evolution of overall concentration field is governed by the Cahn-Hillard equation [5]:

where  is the diffusivity dependent on the phase field . The diffusivity of hydrogen in each phase
is the diffusivity dependent on the phase field . The diffusivity of hydrogen in each phase  could be gotten from experiments or lower scale calculations.
could be gotten from experiments or lower scale calculations.
Conclusion
This proposal detail a multiscale approach for the modeling of hydrogen porosity formation during casting of Al-H system where data are passed from the lower length scale, DFT to the nanoscale, molecular dynamics, and finally to the mesoscale, phase field model. The percent porosity, average pore size, and morphology predicted from the phase field calculations can be further upscaled to an ISV model to predict the mechanical properties (such as yield, ultimate, and fatigue strength) of the cast. This is one of the parameters that will be needed from the molecular dynamics simulations as a function of temperature and composition of hydrogen concentration.
References
- ↑ S. G. Kim, W. T. Kim, and T. Suzuki, “Phase-field model for binary alloys,” Phys. Rev. E, vol. 60, no. 6, pp. 7186–7197, Dec. 1999.
- ↑ N. Moelans, “A quantitative and thermodynamically consistent phase-field interpolation function for multi-phase systems,” Acta Mater., vol. 59, no. 3, pp. 1077–1086, Feb. 2011.
- ↑ L.-Q. Chen and W. Yang, “Computer simulation of the domain dynamics of a quenched system with a large number of nonconserved order parameters: The grain-growth kinetics,” Phys. Rev. B, vol. 50, no. 21, pp. 15752–15756, Dec. 1994.
- ↑ S.M. Allen, J.W. Cahn. Acta Matellurgica 27 (1979) 1085-1095.
- ↑ J.W. Cahn, J.E. Hilliard, The Journal of Chemical Physics 28 (1958) 258.