Getting started for new users
(c.f. general information on the material models at the electronic scale)
Download Quantum Espresso here.
Input files required to run Quantum Espresso
To run Quantum Espresso, all you need is an input file and a pseudopotential and an input script.
Here is an example input script: File:Qe.input.txt
How to run Quantum Espresso
Sample Run with Aluminum
This input file was run using Quantum Espresso using the command assuming that the input file name is 'al.in'
pw.x <scf.in> scf.out& (using only one processor)
mpirun -np 4 pw.x <al.in> al.out (using 4 processors)
In the below input file, the directory to pseudopotentials need to be specified (in this case LDA pseudopotential, Al.pz-n-rrkjus_psl.0.1.UPF is used )Download here.
&CONTROL
calculation = 'scf' ,
outdir = './tmp' ,
pseudo_dir = 'dir/to/pseudopotentials' ,
prefix = 'pwscf' ,
verbosity = 'low' ,
/
&SYSTEM
ibrav = 2,
celldm(1) = 7.6525971195,
nat = 1,
ntyp = 1,
ecutwfc = 30 ,
ecutrho = 120 ,
occupations = 'smearing' ,
degauss = 0.005 ,
smearing = 'marzari-vanderbilt' ,
/
&ELECTRONS
conv_thr = 1d-06 ,
mixing_beta = 0.7d0 ,
/
ATOMIC_SPECIES
Al 26.98150 Al.pz-n-rrkjus_psl.0.1.UPF
ATOMIC_POSITIONS crystal
Al 0.000000000 0.000000000 0.000000000
K_POINTS automatic
8 8 8 0 0 0
|
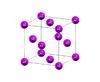 Structure of Aluminum visualized using Xcrysden [ [1]].
The ouput file al.out looks like this
Program PWSCF v.5.1 starts on 4Feb2017 at 19:24:29
This program is part of the open-source Quantum ESPRESSO suite
for quantum simulation of materials; please cite
"P. Giannozzi et al., J. Phys.:Condens. Matter 21 395502 (2009);
URL http://www.quantum-espresso.org",
in publications or presentations arising from this work. More details at
http://www.quantum-espresso.org/quote
Parallel version (MPI), running on 1 processors
Waiting for input...
Reading input from standard input
Current dimensions of program PWSCF are:
Max number of different atomic species (ntypx) = 10
Max number of k-points (npk) = 40000
Max angular momentum in pseudopotentials (lmaxx) = 3
IMPORTANT: XC functional enforced from input :
Exchange-correlation = LDA ( 1 1 0 0 0)
Any further DFT definition will be discarded
Please, verify this is what you really want
Subspace diagonalization in iterative solution of the eigenvalue problem:
a serial algorithm will be used
G-vector sticks info
--------------------
sticks: dense smooth PW G-vecs: dense smooth PW
Sum 241 241 61 2445 2445 331
bravais-lattice index = 2
lattice parameter (alat) = 7.6526 a.u.
unit-cell volume = 112.0383 (a.u.)^3
number of atoms/cell = 1
number of atomic types = 1
number of electrons = 3.00
number of Kohn-Sham states= 6
kinetic-energy cutoff = 30.0000 Ry
charge density cutoff = 120.0000 Ry
convergence threshold = 1.0E-06
mixing beta = 0.7000
number of iterations used = 8 plain mixing
Exchange-correlation = LDA ( 1 1 0 0 0)
celldm(1)= 7.652597 celldm(2)= 0.000000 celldm(3)= 0.000000
celldm(4)= 0.000000 celldm(5)= 0.000000 celldm(6)= 0.000000
crystal axes: (cart. coord. in units of alat)
a(1) = ( -0.500000 0.000000 0.500000 )
a(2) = ( 0.000000 0.500000 0.500000 )
a(3) = ( -0.500000 0.500000 0.000000 )
reciprocal axes: (cart. coord. in units 2 pi/alat)
b(1) = ( -1.000000 -1.000000 1.000000 )
b(2) = ( 1.000000 1.000000 1.000000 )
b(3) = ( -1.000000 1.000000 -1.000000 )
PseudoPot. # 1 for Al read from file:
/home/chaitanya/pseudo/Al.pz-n-rrkjus_psl.0.1.UPF
MD5 check sum: d65347cb939431d5400bc8497a8acd39
Pseudo is Ultrasoft + core correction, Zval = 3.0
Generated using "atomic" code by A. Dal Corso v.5.0.2 svn rev. 9415
Using radial grid of 1135 points, 4 beta functions with:
l(1) = 0
l(2) = 0
l(3) = 1
l(4) = 1
Q(r) pseudized with 0 coefficients
atomic species valence mass pseudopotential
Al 3.00 26.98150 Al( 1.00)
48 Sym. Ops., with inversion, found
Cartesian axes
site n. atom positions (alat units)
1 Al tau( 1) = ( 0.0000000 0.0000000 0.0000000 )
number of k points= 1 Marzari-Vanderbilt smearing, width (Ry)= 0.0050
cart. coord. in units 2pi/alat
k( 1) = ( 0.0000000 0.0000000 0.0000000), wk = 2.0000000
Dense grid: 2445 G-vectors FFT dimensions: ( 20, 20, 20)
Largest allocated arrays est. size (Mb) dimensions
Kohn-Sham Wavefunctions 0.03 Mb ( 331, 6)
NL pseudopotentials 0.04 Mb ( 331, 8)
Each V/rho on FFT grid 0.12 Mb ( 8000)
Each G-vector array 0.02 Mb ( 2445)
G-vector shells 0.00 Mb ( 61)
Largest temporary arrays est. size (Mb) dimensions
Auxiliary wavefunctions 0.12 Mb ( 331, 24)
Each subspace H/S matrix 0.01 Mb ( 24, 24)
Each <psi_i|beta_j> matrix 0.00 Mb ( 8, 6)
Arrays for rho mixing 0.98 Mb ( 8000, 8)
Check: negative/imaginary core charge= -0.000006 0.000000
Initial potential from superposition of free atoms
starting charge 2.99797, renormalised to 3.00000
Starting wfc are 4 randomized atomic wfcs + 2 random wfc
total cpu time spent up to now is 1.8 secs
per-process dynamical memory: 6.3 Mb
Self-consistent Calculation
iteration # 1 ecut= 30.00 Ry beta=0.70
Davidson diagonalization with overlap
ethr = 1.00E-02, avg # of iterations = 6.0
Threshold (ethr) on eigenvalues was too large:
Diagonalizing with lowered threshold
Davidson diagonalization with overlap
ethr = 2.50E-04, avg # of iterations = 4.0
total cpu time spent up to now is 2.0 secs
total energy = -5.22889740 Ry
Harris-Foulkes estimate = -5.22816742 Ry
estimated scf accuracy < 0.00757953 Ry
iteration # 2 ecut= 30.00 Ry beta=0.70
Davidson diagonalization with overlap
ethr = 2.53E-04, avg # of iterations = 1.0
total cpu time spent up to now is 2.0 secs
total energy = -5.22906489 Ry
Harris-Foulkes estimate = -5.22895148 Ry
estimated scf accuracy < 0.00065046 Ry
iteration # 3 ecut= 30.00 Ry beta=0.70
Davidson diagonalization with overlap
ethr = 2.17E-05, avg # of iterations = 1.0
total cpu time spent up to now is 2.1 secs
End of self-consistent calculation
k = 0.0000 0.0000 0.0000 ( 331 PWs) bands (ev):
-3.4045 20.1579 20.1579 21.4744 21.4744 21.4744
the Fermi energy is 20.1412 ev
! total energy = -5.22909765 Ry
Harris-Foulkes estimate = -5.22909744 Ry
estimated scf accuracy < 0.00000061 Ry
The total energy is the sum of the following terms:
one-electron contribution = 3.18844966 Ry
hartree contribution = 0.01500066 Ry
xc contribution = -3.03735564 Ry
ewald contribution = -5.39212484 Ry
smearing contrib. (-TS) = -0.00306748 Ry
convergence has been achieved in 3 iterations
Writing output data file pwscf.save
init_run : 0.97s CPU 1.15s WALL ( 1 calls)
electrons : 0.13s CPU 0.24s WALL ( 1 calls)
Called by init_run:
wfcinit : 0.01s CPU 0.00s WALL ( 1 calls)
potinit : 0.01s CPU 0.02s WALL ( 1 calls)
Called by electrons:
c_bands : 0.04s CPU 0.09s WALL ( 4 calls)
sum_band : 0.04s CPU 0.06s WALL ( 4 calls)
v_of_rho : 0.02s CPU 0.03s WALL ( 4 calls)
newd : 0.03s CPU 0.04s WALL ( 4 calls)
mix_rho : 0.00s CPU 0.00s WALL ( 4 calls)
Called by c_bands:
init_us_2 : 0.01s CPU 0.00s WALL ( 9 calls)
cegterg : 0.04s CPU 0.08s WALL ( 4 calls)
Called by *egterg:
h_psi : 0.02s CPU 0.04s WALL ( 17 calls)
s_psi : 0.00s CPU 0.02s WALL ( 17 calls)
g_psi : 0.00s CPU 0.00s WALL ( 12 calls)
cdiaghg : 0.02s CPU 0.01s WALL ( 15 calls)
Called by h_psi:
add_vuspsi : 0.00s CPU 0.00s WALL ( 17 calls)
General routines
calbec : 0.00s CPU 0.00s WALL ( 21 calls)
fft : 0.02s CPU 0.01s WALL ( 26 calls)
fftw : 0.02s CPU 0.03s WALL ( 174 calls)
davcio : 0.00s CPU 0.00s WALL ( 1 calls)
Parallel routines
fft_scatter : 0.01s CPU 0.00s WALL ( 200 calls)
PWSCF : 1.52s CPU 2.28s WALL
This run was terminated on: 19:24:31 4Feb2017
=------------------------------------------------------------------------------=
JOB DONE.
=------------------------------------------------------------------------------=
|
The total energy can be obtained by usning the command
grep '! total energy' al.out
Another example: Generating a Volume-Energy Curve
|
Description
Source Codes of the Scripts
To see a script click on the link. To download right-click on the link and select "Save Link As".
NOTE: all scripts has been uploaded with .txt extension. When downloading you may want to get rid of it.
By downloading these codes you accept the Mississippi State University's license agreement. Please read the agreement carefully before downloading.
Rasmol source code and documentation for this simply molecular viewer available at rasmol.org
To report bugs, problems or to make comments please use the discussion tab above.
|
